
Как мы использовали WebAssembly для ускорения нашего веб-приложения в 20 раз (пример из практики)
Опубликовано: 2022-03-10Если вы еще не слышали, вот TL;DR: WebAssembly — это новый язык, который работает в браузере вместе с JavaScript. Да все верно. JavaScript больше не единственный язык, который работает в браузере!
Но помимо того, что он «не JavaScript», его отличительной чертой является то, что вы можете компилировать код из таких языков, как C/C++/Rust ( и других! ) в WebAssembly и запускать его в браузере. Поскольку WebAssembly имеет статическую типизацию, использует линейную память и хранится в компактном двоичном формате, он также очень быстр и в конечном итоге может позволить нам выполнять код на «почти исходных» скоростях, то есть на скоростях, близких к вашим. d получить, запустив двоичный файл в командной строке. Возможность использовать существующие инструменты и библиотеки для использования в браузере и связанный с этим потенциал для ускорения — вот две причины, делающие WebAssembly таким привлекательным для Интернета.
До сих пор WebAssembly использовался для самых разных приложений, начиная от игр (например, Doom 3) и заканчивая переносом настольных приложений в Интернет (например, Autocad и Figma). Он даже используется вне браузера, например, как эффективный и гибкий язык для бессерверных вычислений.
Эта статья представляет собой пример использования WebAssembly для ускорения веб-инструмента анализа данных. С этой целью мы возьмем существующий инструмент, написанный на C, который выполняет те же вычисления, скомпилируем его в WebAssembly и используем для замены медленных вычислений JavaScript.
Примечание . В этой статье рассматриваются некоторые сложные темы, такие как компиляция кода C, но не беспокойтесь, если у вас нет опыта в этом; вы по-прежнему сможете следить и понимать, что возможно с WebAssembly.
Задний план
Веб-приложение, с которым мы будем работать, — это fastq.bio, интерактивный веб-инструмент, который предоставляет ученым быстрый предварительный просмотр качества их данных секвенирования ДНК; секвенирование — это процесс, посредством которого мы читаем «буквы» (то есть нуклеотиды) в образце ДНК.
Вот скриншот приложения в действии:
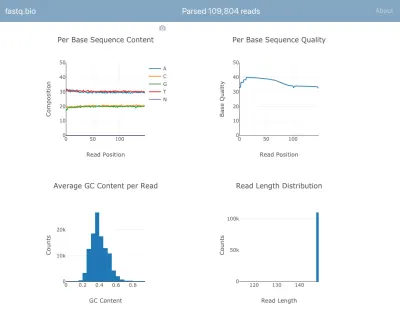
Мы не будем вдаваться в подробности расчетов, но вкратце приведенные выше графики дают ученым представление о том, насколько хорошо прошло секвенирование, и используются для быстрого выявления проблем с качеством данных.
Хотя для создания таких отчетов о контроле качества доступны десятки инструментов командной строки, цель fastq.bio — предоставить интерактивный предварительный просмотр качества данных, не выходя из браузера. Это особенно полезно для ученых, которым не нравится командная строка.
Входные данные для приложения — это обычный текстовый файл, который выводится инструментом секвенирования и содержит список последовательностей ДНК и показатель качества для каждого нуклеотида в последовательностях ДНК. Формат этого файла известен как «FASTQ», отсюда и название fastq.bio.
Если вам интересно узнать о формате FASTQ (необязательно для понимания этой статьи), посетите страницу Википедии, посвященную FASTQ. (Предупреждение: известно, что формат файла FASTQ вызывает фейспалм.)
fastq.bio: реализация JavaScript
В исходной версии fastq.bio пользователь начинает с выбора файла FASTQ на своем компьютере. С помощью объекта File приложение считывает небольшой фрагмент данных, начиная со случайной позиции байта (используя API FileReader). В этом блоке данных мы используем JavaScript для выполнения основных манипуляций со строками и расчета соответствующих показателей. Одна из таких метрик помогает нам отслеживать, сколько A, C, G и T мы обычно видим в каждой позиции фрагмента ДНК.
Как только метрики рассчитаны для этого фрагмента данных, мы отображаем результаты в интерактивном режиме с помощью Plotly.js и переходим к следующему фрагменту в файле. Причина обработки файла небольшими фрагментами заключается в том, чтобы просто улучшить взаимодействие с пользователем: обработка всего файла за один раз заняла бы слишком много времени, поскольку файлы FASTQ обычно имеют размер в сотни гигабайт. Мы обнаружили, что размер фрагмента от 0,5 МБ до 1 МБ сделает приложение более цельным и будет быстрее возвращать информацию пользователю, но это число будет варьироваться в зависимости от деталей вашего приложения и объема вычислений.
Архитектура нашей исходной реализации JavaScript была довольно простой:
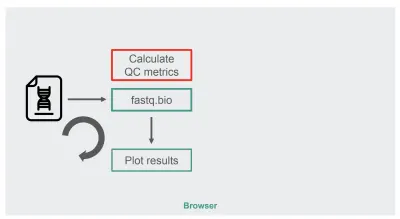
Красное поле — это место, где мы выполняем манипуляции со строками для создания метрик. Этот блок является наиболее ресурсоемкой частью приложения, что, естественно, сделало его хорошим кандидатом для оптимизации во время выполнения с помощью WebAssembly.
fastq.bio: реализация WebAssembly
Чтобы выяснить, можем ли мы использовать WebAssembly для ускорения нашего веб-приложения, мы искали готовый инструмент, который вычисляет показатели контроля качества для файлов FASTQ. В частности, мы искали инструмент, написанный на C/C++/Rust, чтобы его можно было портировать на WebAssembly, и который уже был проверен и которому доверяет научное сообщество.
После некоторых исследований мы решили использовать seqtk, широко используемый инструмент с открытым исходным кодом, написанный на C, который может помочь нам оценить качество данных секвенирования (и чаще используется для управления этими файлами данных).
Прежде чем мы скомпилируем в WebAssembly, давайте сначала рассмотрим, как мы обычно компилируем seqtk в двоичный файл, чтобы запускать его в командной строке. Согласно Makefile, это заклинание gcc , которое вам нужно:
# Compile to binary $ gcc seqtk.c \ -o seqtk \ -O2 \ -lm \ -lzС другой стороны, чтобы скомпилировать seqtk в WebAssembly, мы можем использовать цепочку инструментов Emscripten, которая обеспечивает замену существующих инструментов сборки, чтобы упростить работу в WebAssembly. Если у вас не установлен Emscripten, вы можете загрузить образ докера, который мы подготовили на Dockerhub, в котором есть необходимые вам инструменты (вы также можете установить его с нуля, но обычно это занимает некоторое время):

$ docker pull robertaboukhalil/emsdk:1.38.26 $ docker run -dt --name wasm-seqtk robertaboukhalil/emsdk:1.38.26 Внутри контейнера мы можем использовать компилятор emcc вместо gcc :
# Compile to WebAssembly $ emcc seqtk.c \ -o seqtk.js \ -O2 \ -lm \ -s USE_ZLIB=1 \ -s FORCE_FILESYSTEM=1Как видите, различия между компиляцией в бинарник и WebAssembly минимальны:
- Вместо того, чтобы на выходе был двоичный файл
seqtk, мы просим Emscripten сгенерировать.wasmи.js, которые обрабатывают экземпляр нашего модуля WebAssembly. - Для поддержки библиотеки zlib мы используем флаг
USE_ZLIB; zlib настолько распространен, что уже портирован на WebAssembly, и Emscripten включит его для нас в наш проект. - Мы включаем виртуальную файловую систему Emscripten, которая является POSIX-подобной файловой системой (исходный код здесь), за исключением того, что она работает в оперативной памяти в браузере и исчезает при обновлении страницы (если только вы не сохраните ее состояние в браузере с помощью IndexedDB, но это для другой статьи).
Почему виртуальная файловая система? Чтобы ответить на этот вопрос, давайте сравним, как мы будем вызывать seqtk в командной строке и как использовать JavaScript для вызова скомпилированного модуля WebAssembly:
# On the command line $ ./seqtk fqchk data.fastq # In the browser console > Module.callMain(["fqchk", "data.fastq"]) Иметь доступ к виртуальной файловой системе очень важно, потому что это означает, что нам не нужно переписывать seqtk для обработки строковых входных данных вместо путей к файлам. Мы можем смонтировать кусок данных как файл data.fastq в виртуальной файловой системе и просто вызвать для него функцию main() seqtk.
С seqtk, скомпилированным в WebAssembly, вот новая архитектура fastq.bio:
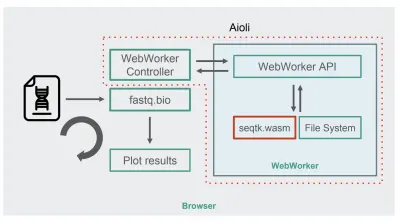
Как показано на диаграмме, вместо выполнения вычислений в основном потоке браузера мы используем WebWorkers, которые позволяют нам выполнять наши вычисления в фоновом потоке и избегать негативного влияния на скорость отклика браузера. В частности, контроллер WebWorker запускает Worker и управляет связью с основным потоком. На стороне Worker API выполняет полученные запросы.
Затем мы можем попросить Worker запустить команду seqtk для файла, который мы только что смонтировали. Когда seqtk завершает работу, Worker отправляет результат обратно в основной поток через Promise. Получив сообщение, основной поток использует полученные выходные данные для обновления диаграмм. Как и в версии JavaScript, мы обрабатываем файлы фрагментами и обновляем визуализацию на каждой итерации.
Оптимизация производительности
Чтобы оценить, принесло ли пользу использование WebAssembly, мы сравнили реализации JavaScript и WebAssembly, используя метрику того, сколько операций чтения мы можем обработать в секунду. Мы игнорируем время, необходимое для создания интерактивных графиков, поскольку обе реализации используют для этой цели JavaScript.
Из коробки мы уже видим ускорение ~9X:
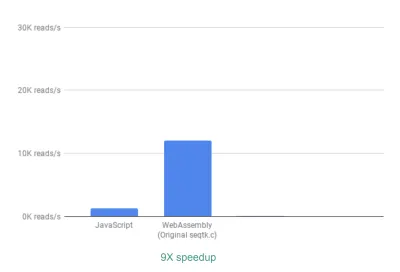
Это уже очень хорошо, учитывая, что этого было относительно легко достичь (то есть, когда вы понимаете WebAssembly!).
Затем мы заметили, что, хотя seqtk выводит множество обычно полезных метрик контроля качества, многие из этих метрик на самом деле не используются и не отображаются нашим приложением. Удалив часть вывода для метрик, которые нам не нужны, мы смогли увидеть еще большее ускорение в 13 раз:
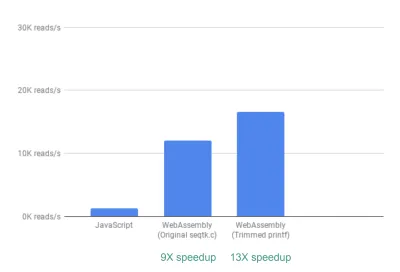
Это снова большое улучшение, учитывая, как легко его было добиться — буквально закомментировав операторы printf, которые не были нужны.
Наконец, есть еще одно улучшение, которое мы рассмотрели. Пока что способ, которым fastq.bio получает интересующие метрики, заключается в вызове двух разных функций C, каждая из которых вычисляет свой набор метрик. В частности, одна функция возвращает информацию в виде гистограммы (т. е. списка значений, которые мы объединяем в диапазоны), тогда как другая функция возвращает информацию в виде функции положения последовательности ДНК. К сожалению, это означает, что один и тот же фрагмент файла читается дважды, что необязательно.
Итак, мы объединили код двух функций в одну, хотя и беспорядочную, функцию (даже не освежив мой C!). Поскольку два вывода имеют разное количество столбцов, мы немного потрудились на стороне JavaScript, чтобы разделить их. Но оно того стоило: это позволило нам добиться более чем 20-кратного ускорения!
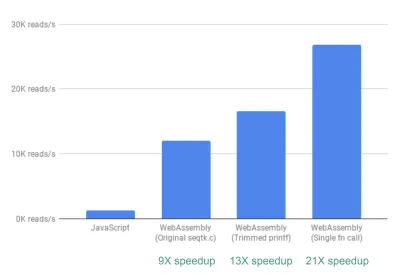
Слово предостережения
Сейчас самое время сделать предостережение. Не ожидайте, что вы всегда получите 20-кратное ускорение при использовании WebAssembly. Вы можете получить ускорение только в 2 раза или на 20%. Или вы можете столкнуться с замедлением, если загружаете в память очень большие файлы или требуете интенсивного обмена данными между WebAssembly и JavaScript.
Заключение
Короче говоря, мы видели, что замена медленных вычислений JavaScript вызовами скомпилированного WebAssembly может привести к значительному ускорению. Поскольку код, необходимый для этих вычислений, уже существовал на C, мы получили дополнительное преимущество от повторного использования надежного инструмента. Как мы уже упоминали, WebAssembly не всегда будет подходящим инструментом для работы ( вздох! ), так что используйте его с умом.
Дальнейшее чтение
- «Повысьте уровень с помощью WebAssembly», Роберт Абукхалил
Практическое руководство по созданию приложений WebAssembly. - Айоли (на GitHub)
Фреймворк для создания быстрых веб-инструментов геномики. - Исходный код fastq.bio (на GitHub)
Интерактивный веб-инструмент для контроля качества данных секвенирования ДНК. - «Сокращенное мультяшное введение в WebAssembly», Лин Кларк