
Como usamos o WebAssembly para acelerar nosso aplicativo da Web em 20 vezes (estudo de caso)
Publicados: 2022-03-10Se você ainda não ouviu, aqui está o TL;DR: WebAssembly é uma nova linguagem que roda no navegador junto com o JavaScript. Sim está certo. JavaScript não é mais a única linguagem que roda no navegador!
Mas além de ser “não JavaScript”, seu diferencial é que você pode compilar código de linguagens como C/C++/Rust ( e mais! ) para WebAssembly e executá-los no navegador. Como o WebAssembly é tipado estaticamente, usa uma memória linear e é armazenado em um formato binário compacto, também é muito rápido e pode eventualmente nos permitir executar código em velocidades “quase nativas”, ou seja, em velocidades próximas ao que você d get executando o binário na linha de comando. A capacidade de aproveitar as ferramentas e bibliotecas existentes para uso no navegador e o potencial associado de aceleração são dois motivos que tornam o WebAssembly tão atraente para a web.
Até agora, o WebAssembly tem sido usado para todos os tipos de aplicativos, desde jogos (por exemplo, Doom 3), até portar aplicativos de desktop para a web (por exemplo, Autocad e Figma). Ele é usado até mesmo fora do navegador, por exemplo, como uma linguagem eficiente e flexível para computação sem servidor.
Este artigo é um estudo de caso sobre o uso do WebAssembly para acelerar uma ferramenta web de análise de dados. Para esse fim, vamos pegar uma ferramenta existente escrita em C que executa os mesmos cálculos, compilá-la para o WebAssembly e usá-la para substituir cálculos lentos de JavaScript.
Observação : este artigo aborda alguns tópicos avançados, como compilar código C, mas não se preocupe se você não tiver experiência com isso; você ainda poderá acompanhar e ter uma noção do que é possível com o WebAssembly.
Fundo
O aplicativo web com o qual trabalharemos é o fastq.bio, uma ferramenta web interativa que fornece aos cientistas uma visualização rápida da qualidade de seus dados de sequenciamento de DNA; sequenciamento é o processo pelo qual lemos as “letras” (ou seja, nucleotídeos) em uma amostra de DNA.
Aqui está uma captura de tela do aplicativo em ação:
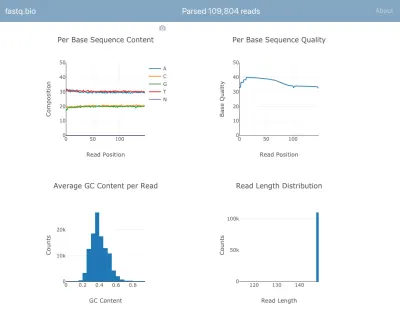
Não entraremos nos detalhes dos cálculos, mas em poucas palavras, os gráficos acima fornecem aos cientistas uma noção de quão bem o sequenciamento foi e são usados para identificar rapidamente problemas de qualidade de dados.
Embora existam dezenas de ferramentas de linha de comando disponíveis para gerar esses relatórios de controle de qualidade, o objetivo do fastq.bio é fornecer uma visualização interativa da qualidade dos dados sem sair do navegador. Isso é especialmente útil para cientistas que não se sentem à vontade com a linha de comando.
A entrada para o aplicativo é um arquivo de texto simples que é gerado pelo instrumento de sequenciamento e contém uma lista de sequências de DNA e um índice de qualidade para cada nucleotídeo nas sequências de DNA. O formato desse arquivo é conhecido como “FASTQ”, daí o nome fastq.bio.
Se você está curioso sobre o formato FASTQ (não é necessário para entender este artigo), confira a página do FASTQ na Wikipedia. (Aviso: O formato de arquivo FASTQ é conhecido no campo por induzir facepalms.)
fastq.bio: A implementação do JavaScript
Na versão original do fastq.bio, o usuário começa selecionando um arquivo FASTQ de seu computador. Com o objeto File , o aplicativo lê um pequeno pedaço de dados começando em uma posição de byte aleatória (usando a API FileReader). Nesse bloco de dados, usamos JavaScript para realizar manipulações básicas de strings e calcular métricas relevantes. Uma dessas métricas nos ajuda a rastrear quantos A, C, G e T que normalmente vemos em cada posição ao longo de um fragmento de DNA.
Depois que as métricas são calculadas para esse bloco de dados, plotamos os resultados interativamente com Plotly.js e passamos para o próximo bloco no arquivo. A razão para processar o arquivo em pequenos pedaços é simplesmente melhorar a experiência do usuário: processar o arquivo inteiro de uma vez levaria muito tempo, porque os arquivos FASTQ geralmente têm centenas de gigabytes. Descobrimos que um tamanho de bloco entre 0,5 MB e 1 MB tornaria o aplicativo mais transparente e retornaria informações ao usuário mais rapidamente, mas esse número varia de acordo com os detalhes do seu aplicativo e o peso dos cálculos.
A arquitetura de nossa implementação JavaScript original era bastante direta:
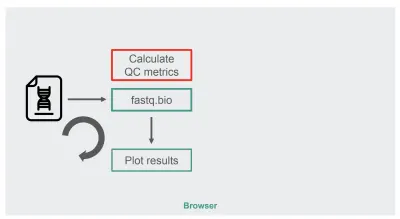
A caixa em vermelho é onde fazemos as manipulações de strings para gerar as métricas. Essa caixa é a parte mais intensiva de computação do aplicativo, o que naturalmente a tornou uma boa candidata para otimização de tempo de execução com WebAssembly.
fastq.bio: A implementação do WebAssembly
Para explorar se poderíamos aproveitar o WebAssembly para acelerar nosso aplicativo da Web, procuramos uma ferramenta pronta para uso que calcula métricas de CQ em arquivos FASTQ. Especificamente, buscou-se uma ferramenta escrita em C/C++/Rust para que fosse passível de portabilidade para WebAssembly, e que já fosse validada e confiável pela comunidade científica.
Após algumas pesquisas, decidimos usar o seqtk, uma ferramenta de código aberto comumente usada escrita em C que pode nos ajudar a avaliar a qualidade dos dados de sequenciamento (e geralmente é usada para manipular esses arquivos de dados).
Antes de compilar para o WebAssembly, vamos primeiro considerar como normalmente compilaríamos o seqtk para binário para executá-lo na linha de comando. De acordo com o Makefile, este é o encantamento gcc que você precisa:
# Compile to binary $ gcc seqtk.c \ -o seqtk \ -O2 \ -lm \ -lzPor outro lado, para compilar o seqtk para o WebAssembly, podemos usar a cadeia de ferramentas Emscripten, que fornece substituições para ferramentas de compilação existentes para facilitar o trabalho no WebAssembly. Se você não tem o Emscripten instalado, você pode baixar uma imagem docker que preparamos no Dockerhub que tem as ferramentas que você precisa (você também pode instalá-lo do zero, mas isso geralmente demora um pouco):

$ docker pull robertaboukhalil/emsdk:1.38.26 $ docker run -dt --name wasm-seqtk robertaboukhalil/emsdk:1.38.26 Dentro do container, podemos usar o compilador emcc como substituto do gcc :
# Compile to WebAssembly $ emcc seqtk.c \ -o seqtk.js \ -O2 \ -lm \ -s USE_ZLIB=1 \ -s FORCE_FILESYSTEM=1Como você pode ver, as diferenças entre compilar para binário e WebAssembly são mínimas:
- Em vez da saída ser o arquivo binário
seqtk, pedimos ao Emscripten para gerar um.wasme um.jsque tratam da instanciação do nosso módulo WebAssembly - Para suportar a biblioteca zlib, usamos o sinalizador
USE_ZLIB; zlib é tão comum que já foi portado para o WebAssembly, e o Emscripten irá incluí-lo para nós em nosso projeto - Habilitamos o sistema de arquivos virtual do Emscripten, que é um sistema de arquivos semelhante ao POSIX (código-fonte aqui), exceto que ele é executado na RAM no navegador e desaparece quando você atualiza a página (a menos que você salve seu estado no navegador usando IndexedDB, mas isso é para outro artigo).
Por que um sistema de arquivos virtual? Para responder a isso, vamos comparar como chamaríamos seqtk na linha de comando versus usar JavaScript para chamar o módulo WebAssembly compilado:
# On the command line $ ./seqtk fqchk data.fastq # In the browser console > Module.callMain(["fqchk", "data.fastq"]) Ter acesso a um sistema de arquivos virtual é poderoso porque significa que não precisamos reescrever seqtk para manipular entradas de string em vez de caminhos de arquivo. Podemos montar um pedaço de dados como o arquivo data.fastq no sistema de arquivos virtual e simplesmente chamar a função main() do seqtk nele.
Com o seqtk compilado para o WebAssembly, aqui está a nova arquitetura fastq.bio:
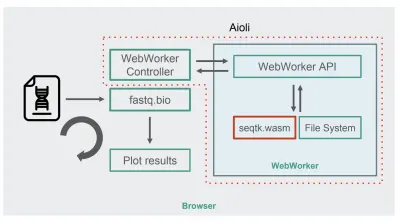
Conforme mostrado no diagrama, em vez de executar os cálculos no thread principal do navegador, usamos WebWorkers, que nos permitem executar nossos cálculos em um thread em segundo plano e evitar afetar negativamente a capacidade de resposta do navegador. Especificamente, o controlador WebWorker inicia o Worker e gerencia a comunicação com o thread principal. Do lado do Worker, uma API executa as solicitações que recebe.
Podemos então pedir ao Worker para executar um comando seqtk no arquivo que acabamos de montar. Quando o seqtk termina de ser executado, o Worker envia o resultado de volta ao thread principal por meio de um Promise. Depois de receber a mensagem, o encadeamento principal usa a saída resultante para atualizar os gráficos. Semelhante à versão JavaScript, processamos os arquivos em partes e atualizamos as visualizações a cada iteração.
Otimização de performance
Para avaliar se o uso do WebAssembly foi bom, comparamos as implementações JavaScript e WebAssembly usando a métrica de quantas leituras podemos processar por segundo. Ignoramos o tempo que leva para gerar gráficos interativos, pois ambas as implementações usam JavaScript para essa finalidade.
Fora da caixa, já vemos uma aceleração de ~ 9X:
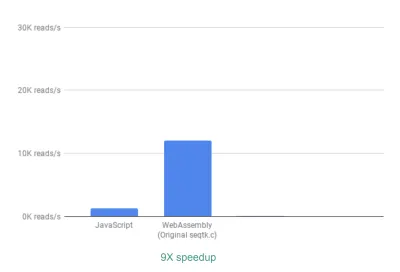
Isso já é muito bom, já que foi relativamente fácil de conseguir (isto é, uma vez que você entenda o WebAssembly!).
Em seguida, notamos que, embora o seqtk gere muitas métricas de controle de qualidade geralmente úteis, muitas dessas métricas não são realmente usadas ou representadas graficamente por nosso aplicativo. Ao remover parte da saída para as métricas que não precisávamos, conseguimos ver uma aceleração ainda maior de 13X:
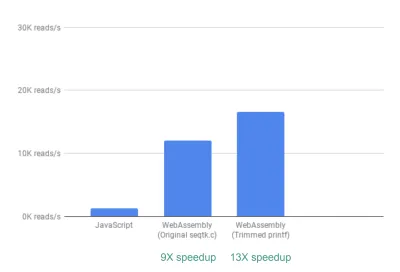
Novamente, isso é uma grande melhoria, considerando a facilidade com que foi alcançado - literalmente comentando as instruções printf que não eram necessárias.
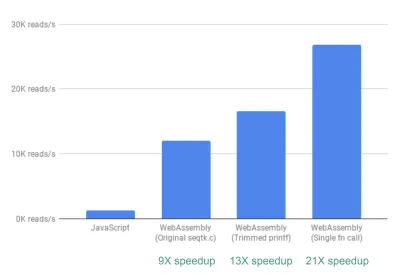
Finalmente, há mais uma melhoria que analisamos. Até agora, a forma como o fastq.bio obtém as métricas de interesse é chamando duas funções C diferentes, cada uma das quais calcula um conjunto diferente de métricas. Especificamente, uma função retorna informações na forma de um histograma (ou seja, uma lista de valores que agrupamos em intervalos), enquanto a outra função retorna informações em função da posição da sequência de DNA. Infelizmente, isso significa que o mesmo pedaço de arquivo é lido duas vezes, o que é desnecessário.
Então nós mesclamos o código para as duas funções em uma – embora confusa – função (sem ter que retocar meu C!). Como as duas saídas têm números diferentes de colunas, fizemos algumas discussões no lado do JavaScript para separar as duas. Mas valeu a pena: isso nos permitiu atingir uma aceleração >20X!
Uma palavra de cautela
Agora seria um bom momento para uma ressalva. Não espere obter sempre uma aceleração de 20 vezes ao usar o WebAssembly. Você pode obter apenas uma aceleração de 2X ou uma aceleração de 20%. Ou você pode ficar mais lento se carregar arquivos muito grandes na memória ou exigir muita comunicação entre o WebAssembly e o JavaScript.
Conclusão
Resumindo, vimos que substituir cálculos JavaScript lentos por chamadas para WebAssembly compilado pode levar a acelerações significativas. Como o código necessário para esses cálculos já existia em C, tivemos o benefício adicional de reutilizar uma ferramenta confiável. Como também mencionamos, o WebAssembly nem sempre será a ferramenta certa para o trabalho ( suspiro! ), então use-o com sabedoria.
Leitura adicional
- “Suba de nível com o WebAssembly”, Robert Aboukhalil
Um guia prático para construir aplicativos WebAssembly. - Aioli (no GitHub)
Um framework para construir ferramentas web de genômica rápida. - código fonte fastq.bio (no GitHub)
Uma ferramenta web interativa para controle de qualidade de dados de sequenciamento de DNA. - “Uma introdução resumida dos desenhos animados ao WebAssembly”, Lin Clark