
Jak wykorzystaliśmy WebAssembly do 20-krotnego przyspieszenia naszej aplikacji internetowej (studium przypadku)
Opublikowany: 2022-03-10Jeśli nie słyszałeś, oto TL; DR: WebAssembly to nowy język, który działa w przeglądarce wraz z JavaScript. Tak to prawda. JavaScript nie jest już jedynym językiem działającym w przeglądarce!
Ale poza tym, że jest „nie JavaScript”, jego wyróżnikiem jest to, że możesz kompilować kod z języków takich jak C/C++/Rust ( i nie tylko! ) do WebAssembly i uruchamiać go w przeglądarce. Ponieważ WebAssembly jest statycznie typowany, wykorzystuje pamięć liniową i jest przechowywany w kompaktowym formacie binarnym, jest również bardzo szybki i może ostatecznie pozwolić nam na uruchamianie kodu z „prawie natywnymi” prędkościami, tj. z prędkościami zbliżonymi do Twoich d uzyskać, uruchamiając plik binarny w wierszu poleceń. Możliwość wykorzystania istniejących narzędzi i bibliotek do użycia w przeglądarce i związany z tym potencjał przyspieszenia to dwa powody, które sprawiają, że WebAssembly jest tak atrakcyjny w sieci.
Do tej pory WebAssembly był używany do wszelkiego rodzaju aplikacji, począwszy od gier (np. Doom 3), po przenoszenie aplikacji desktopowych do sieci (np. Autocad i Figma). Jest używany nawet poza przeglądarką, na przykład jako wydajny i elastyczny język do przetwarzania bezserwerowego.
Ten artykuł jest studium przypadku wykorzystania WebAssembly do przyspieszenia narzędzia internetowego do analizy danych. W tym celu weźmiemy istniejące narzędzie napisane w C, które wykonuje te same obliczenia, skompilujemy je do WebAssembly i użyjemy do zastąpienia powolnych obliczeń JavaScript.
Uwaga : ten artykuł zagłębia się w niektóre zaawansowane tematy, takie jak kompilacja kodu C, ale nie martw się, jeśli nie masz z tym doświadczenia; nadal będziesz mógł podążać dalej i zorientować się, co jest możliwe dzięki WebAssembly.
Tło
Aplikacja internetowa, z którą będziemy pracować, to fastq.bio, interaktywne narzędzie internetowe, które zapewnia naukowcom szybki podgląd jakości danych sekwencjonowania DNA; sekwencjonowanie to proces, w którym odczytujemy „litery” (tj. nukleotydy) w próbce DNA.
Oto zrzut ekranu aplikacji w akcji:
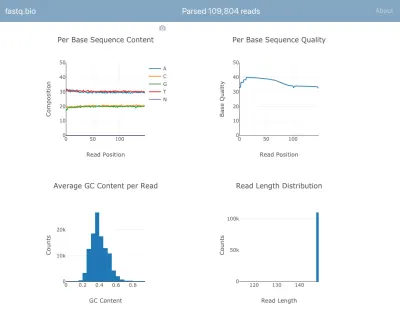
Nie będziemy wchodzić w szczegóły obliczeń, ale w skrócie, powyższe wykresy dostarczają naukowcom informacji o tym, jak dobrze przebiegło sekwencjonowanie i są wykorzystywane do szybkiego identyfikowania problemów z jakością danych.
Chociaż dostępne są dziesiątki narzędzi wiersza poleceń do generowania takich raportów kontroli jakości, celem fastq.bio jest zapewnienie interaktywnego podglądu jakości danych bez opuszczania przeglądarki. Jest to szczególnie przydatne dla naukowców, którzy nie są zaznajomieni z wierszem poleceń.
Dane wejściowe do aplikacji to zwykły plik tekstowy, który jest wysyłany przez instrument do sekwencjonowania i zawiera listę sekwencji DNA oraz wynik jakości dla każdego nukleotydu w sekwencjach DNA. Format tego pliku jest znany jako „FASTQ”, stąd nazwa fastq.bio.
Jeśli interesuje Cię format FASTQ (nie jest to konieczne do zrozumienia tego artykułu), sprawdź stronę Wikipedii dla FASTQ. (Ostrzeżenie: format pliku FASTQ jest znany w terenie, aby wywoływać facepalms.)
fastq.bio: Implementacja JavaScript
W oryginalnej wersji fastq.bio użytkownik zaczyna od wybrania pliku FASTQ ze swojego komputera. Za pomocą obiektu File aplikacja odczytuje niewielki fragment danych, zaczynając od losowej pozycji bajtu (przy użyciu interfejsu API FileReader). W tym kawałku danych używamy JavaScript do wykonywania podstawowych operacji na ciągach znaków i obliczania odpowiednich metryk. Jedna z takich metryk pomaga nam śledzić, ile A, C, G i T zazwyczaj widzimy w każdej pozycji we fragmencie DNA.
Po obliczeniu metryk dla tego fragmentu danych interaktywnie wykreślamy wyniki za pomocą Plotly.js i przechodzimy do następnego fragmentu w pliku. Powodem przetwarzania pliku w małych porcjach jest po prostu poprawa komfortu użytkownika: przetwarzanie całego pliku na raz zajęłoby zbyt dużo czasu, ponieważ pliki FASTQ mają zazwyczaj setki gigabajtów. Odkryliśmy, że rozmiar fragmentu między 0,5 MB a 1 MB sprawi, że aplikacja będzie bardziej płynna i szybciej zwróci informacje użytkownikowi, ale liczba ta będzie się różnić w zależności od szczegółów aplikacji i intensywności obliczeń.
Architektura naszej oryginalnej implementacji JavaScript była dość prosta:
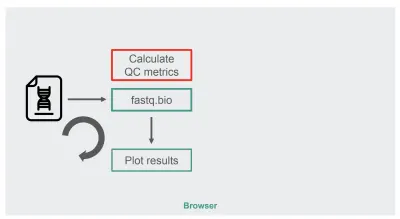
Czerwone pole to miejsce, w którym wykonujemy manipulacje ciągami, aby wygenerować metryki. To pudełko jest bardziej intensywną obliczeniowo częścią aplikacji, co naturalnie czyniło ją dobrym kandydatem do optymalizacji środowiska uruchomieniowego za pomocą WebAssembly.
fastq.bio: Implementacja WebAssembly
Aby sprawdzić, czy możemy wykorzystać WebAssembly do przyspieszenia naszej aplikacji internetowej, szukaliśmy gotowego narzędzia, które oblicza wskaźniki QC w plikach FASTQ. W szczególności szukaliśmy narzędzia napisanego w C/C++/Rust, aby można je było przenieść do WebAssembly, a także takiego, które zostało już sprawdzone i zaufane przez społeczność naukową.
Po kilku badaniach zdecydowaliśmy się na seqtk, powszechnie używane narzędzie o otwartym kodzie źródłowym napisane w C, które może pomóc nam ocenić jakość danych sekwencjonowania (i jest bardziej ogólnie używane do manipulowania tymi plikami danych).
Zanim skompilujemy do WebAssembly, zastanówmy się najpierw, jak normalnie skompilowalibyśmy seqtk do pliku binarnego, aby uruchomić go w wierszu poleceń. Według Makefile jest to inkantacja gcc , której potrzebujesz:
# Compile to binary $ gcc seqtk.c \ -o seqtk \ -O2 \ -lm \ -lzZ drugiej strony, aby skompilować seqtk do WebAssembly, możemy użyć łańcucha narzędzi Emscripten, który zapewnia zamienniki dla istniejących narzędzi do budowania, aby ułatwić pracę w WebAssembly. Jeśli nie masz zainstalowanego Emscripten, możesz pobrać obraz dockera, który przygotowaliśmy na Dockerhub, który zawiera potrzebne narzędzia (możesz też zainstalować go od zera, ale zwykle zajmuje to trochę czasu):

$ docker pull robertaboukhalil/emsdk:1.38.26 $ docker run -dt --name wasm-seqtk robertaboukhalil/emsdk:1.38.26 Wewnątrz kontenera możemy użyć kompilatora emcc jako zamiennika gcc :
# Compile to WebAssembly $ emcc seqtk.c \ -o seqtk.js \ -O2 \ -lm \ -s USE_ZLIB=1 \ -s FORCE_FILESYSTEM=1Jak widać, różnice między kompilacją do binarnego a WebAssembly są minimalne:
- Zamiast wyjścia będącego plikiem binarnym
seqtk, prosimy Emscripten o wygenerowanie.wasmi.js, które obsługują tworzenie instancji naszego modułu WebAssembly - Do obsługi biblioteki zlib używamy flagi
USE_ZLIB; zlib jest tak powszechny, że został już przeniesiony do WebAssembly, a Emscripten uwzględni go dla nas w naszym projekcie - Włączamy wirtualny system plików Emscripten, który jest systemem plików podobnym do POSIX (tutaj kod źródłowy), z wyjątkiem tego, że działa on w pamięci RAM w przeglądarce i znika po odświeżeniu strony (chyba że zapiszesz jego stan w przeglądarce za pomocą IndexedDB, ale tak jest na inny artykuł).
Dlaczego wirtualny system plików? Aby odpowiedzieć na to pytanie, porównajmy, jak wywołalibyśmy seqtk w wierszu poleceń z użyciem JavaScript do wywołania skompilowanego modułu WebAssembly:
# On the command line $ ./seqtk fqchk data.fastq # In the browser console > Module.callMain(["fqchk", "data.fastq"]) Posiadanie dostępu do wirtualnego systemu plików jest potężne, ponieważ oznacza, że nie musimy przepisywać seqtk, aby obsługiwał dane wejściowe zamiast ścieżek plików. Możemy zamontować porcję danych jako plik data.fastq w wirtualnym systemie plików i po prostu wywołać na nim funkcję main() seqtk.
Dzięki seqtk skompilowanym do WebAssembly, oto nowa architektura fastq.bio:
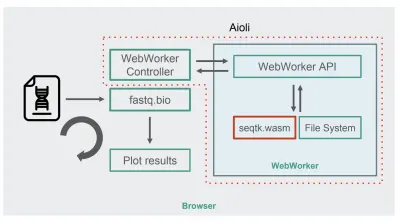
Jak pokazano na diagramie, zamiast uruchamiać obliczenia w głównym wątku przeglądarki, korzystamy z WebWorkerów, które pozwalają nam wykonywać obliczenia w wątku w tle i uniknąć negatywnego wpływu na responsywność przeglądarki. W szczególności kontroler WebWorker uruchamia Worker i zarządza komunikacją z głównym wątkiem. Po stronie pracownika API wykonuje otrzymane żądania.
Następnie możemy poprosić pracownika, aby uruchomił polecenie seqtk na pliku, który właśnie zamontowaliśmy. Kiedy seqtk kończy działanie, Worker wysyła wynik z powrotem do głównego wątku poprzez Promise. Po odebraniu wiadomości wątek główny wykorzystuje wynikowe dane wyjściowe do aktualizacji wykresów. Podobnie jak w wersji JavaScript, przetwarzamy pliki porcjami i aktualizujemy wizualizacje w każdej iteracji.
Optymalizacja wydajności
Aby ocenić, czy użycie WebAssembly dało coś dobrego, porównujemy implementacje JavaScript i WebAssembly, używając metryki liczby odczytów, które możemy przetworzyć na sekundę. Ignorujemy czas potrzebny na generowanie interaktywnych wykresów, ponieważ obie implementacje używają do tego celu JavaScript.
Po wyjęciu z pudełka już widzimy przyspieszenie ~9X:
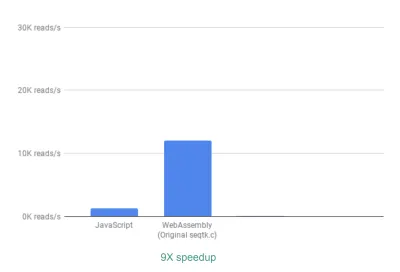
To już bardzo dobrze, biorąc pod uwagę, że było to stosunkowo łatwe do osiągnięcia (to znaczy, kiedy zrozumiesz WebAssembly!).
Następnie zauważyliśmy, że chociaż seqtk generuje wiele ogólnie przydatnych metryk kontroli jakości, wiele z tych metryk nie jest w rzeczywistości używanych ani przedstawianych na wykresach przez naszą aplikację. Usuwając niektóre dane wyjściowe dla metryk, których nie potrzebowaliśmy, byliśmy w stanie zobaczyć jeszcze większe przyspieszenie 13X:
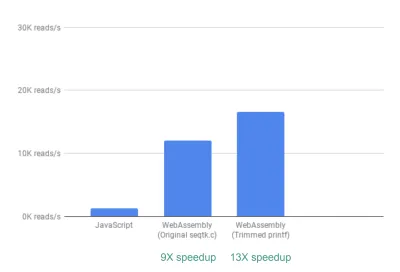
To znowu wielka poprawa, biorąc pod uwagę, jak łatwo było to osiągnąć — poprzez dosłowne komentowanie niepotrzebnych instrukcji printf.
Na koniec przyjrzeliśmy się jeszcze jednej poprawie. Jak dotąd, sposób, w jaki fastq.bio uzyskuje interesujące metryki, polega na wywołaniu dwóch różnych funkcji C, z których każda oblicza inny zestaw metryk. W szczególności jedna funkcja zwraca informacje w postaci histogramu (tj. listy wartości, które łączymy w zakresy), podczas gdy druga funkcja zwraca informacje w postaci funkcji pozycji sekwencji DNA. Niestety oznacza to, że ten sam fragment pliku jest odczytywany dwukrotnie, co nie jest konieczne.
Połączyliśmy więc kod dla dwóch funkcji w jedną — choć niechlujną — funkcję (bez konieczności odświeżenia mojego C!). Ponieważ oba wyjścia mają różną liczbę kolumn, pokłóciliśmy się po stronie JavaScript, aby je rozplątać. Ale było warto: dzięki temu uzyskaliśmy przyspieszenie >20X!
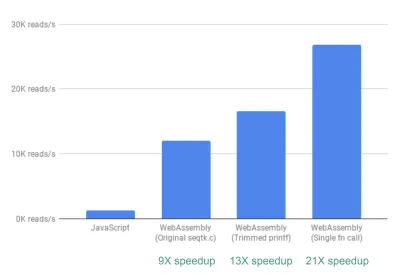
Słowo ostrzeżenia
Teraz byłby dobry czas na zastrzeżenie. Nie oczekuj, że zawsze uzyskasz 20-krotne przyspieszenie podczas korzystania z WebAssembly. Możesz uzyskać tylko przyspieszenie 2X lub przyspieszenie 20%. Lub możesz spowolnić, jeśli ładujesz bardzo duże pliki do pamięci lub potrzebujesz dużo komunikacji między WebAssembly a JavaScriptem.
Wniosek
Krótko mówiąc, widzieliśmy, że zastąpienie powolnych obliczeń JavaScript wywołaniami skompilowanego WebAssembly może prowadzić do znacznego przyspieszenia. Ponieważ kod potrzebny do tych obliczeń istniał już w C, otrzymaliśmy dodatkową korzyść z ponownego użycia zaufanego narzędzia. Jak już wspomnieliśmy, WebAssembly nie zawsze będzie odpowiednim narzędziem do pracy ( sapnięcie! ), więc używaj go mądrze.
Dalsza lektura
- „Podnieś poziom dzięki WebAssembly”, Robert Aboukhalil
Praktyczny przewodnik po budowaniu aplikacji WebAssembly. - Aioli (na GitHubie)
Ramy do tworzenia szybkich narzędzi internetowych genomiki. - Kod źródłowy fastq.bio (na GitHub)
Interaktywne narzędzie internetowe do kontroli jakości danych sekwencjonowania DNA. - „Skrócone wprowadzenie kreskówek do WebAssembly”, Lin Clark