
WebAssembly를 사용하여 웹 앱 속도를 20배 향상시킨 방법(사례 연구)
게시 됨: 2022-03-10아직 듣지 못했다면 다음이 핵심입니다. DR: WebAssembly는 JavaScript와 함께 브라우저에서 실행되는 새로운 언어입니다. 네, 맞습니다. JavaScript는 더 이상 브라우저에서 실행되는 유일한 언어가 아닙니다!
그러나 단순히 "JavaScript가 아님"을 넘어 C/C++/Rust( 및 그 이상! )와 같은 언어의 코드를 WebAssembly로 컴파일하고 브라우저에서 실행할 수 있다는 점에서 구별됩니다. WebAssembly는 정적으로 유형이 지정되고 선형 메모리를 사용하며 컴팩트 바이너리 형식으로 저장되기 때문에 매우 빠르며 결국에는 "네이티브에 가까운" 속도로 코드를 실행할 수 있습니다. d 명령줄에서 바이너리를 실행하여 얻습니다. 브라우저에서 사용하기 위해 기존 도구와 라이브러리를 활용할 수 있는 능력과 이와 관련된 속도 향상 가능성은 WebAssembly를 웹용으로 매우 매력적으로 만드는 두 가지 이유입니다.
지금까지 WebAssembly는 게임(예: Doom 3)에서 데스크톱 응용 프로그램을 웹으로 포팅(예: Autocad 및 Figma)에 이르기까지 모든 종류의 응용 프로그램에 사용되었습니다. 예를 들어 서버리스 컴퓨팅을 위한 효율적이고 유연한 언어로 브라우저 외부에서도 사용됩니다.
이 기사는 WebAssembly를 사용하여 데이터 분석 웹 도구의 속도를 높이는 사례 연구입니다. 이를 위해 동일한 계산을 수행하는 C로 작성된 기존 도구를 가져와 WebAssembly로 컴파일하고 느린 JavaScript 계산을 대체하는 데 사용할 것입니다.
참고 : 이 기사에서는 C 코드 컴파일과 같은 일부 고급 주제에 대해 자세히 설명하지만, 이에 대한 경험이 없더라도 걱정하지 마십시오. 계속해서 WebAssembly로 가능한 것에 대해 이해하고 따라갈 수 있을 것입니다.
배경
우리가 작업할 웹 앱은 과학자들에게 DNA 시퀀싱 데이터의 품질에 대한 빠른 미리보기를 제공하는 대화형 웹 도구인 fastq.bio입니다. 시퀀싱은 DNA 샘플에서 "문자"(즉, 뉴클레오타이드)를 읽는 과정입니다.
다음은 작동 중인 애플리케이션의 스크린샷입니다.
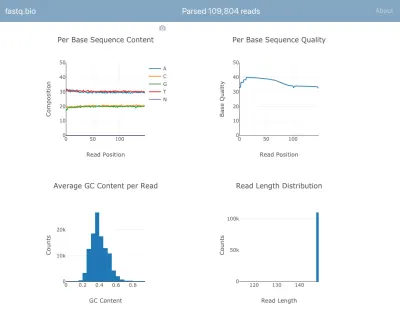
계산의 세부 사항은 다루지 않겠지만 간단히 말해서 위의 플롯은 과학자들에게 시퀀싱이 얼마나 잘 진행되었는지에 대한 감각을 제공하고 데이터 품질 문제를 한 눈에 식별하는 데 사용됩니다.
이러한 품질 관리 보고서를 생성하는 데 사용할 수 있는 수십 가지 명령줄 도구가 있지만 fastq.bio의 목표는 브라우저를 벗어나지 않고 데이터 품질에 대한 대화식 미리 보기를 제공하는 것입니다. 이것은 명령줄에 익숙하지 않은 과학자에게 특히 유용합니다.
앱에 대한 입력은 시퀀싱 기기에서 출력되는 일반 텍스트 파일로, DNA 시퀀스 목록과 DNA 시퀀스의 각 뉴클레오티드에 대한 품질 점수가 포함되어 있습니다. 해당 파일의 형식은 "FASTQ"로 알려져 있으므로 이름이 fastq.bio입니다.
FASTQ 형식에 대해 궁금하다면(이 기사를 이해하는 데 필요하지 않음) FASTQ에 대한 Wikipedia 페이지를 확인하십시오. (경고: FASTQ 파일 형식은 페이스팜을 유도하는 것으로 현장에 알려져 있습니다.)
fastq.bio: 자바스크립트 구현
fastq.bio의 원본 버전에서 사용자는 컴퓨터에서 FASTQ 파일을 선택하여 시작합니다. File 객체를 사용하여 앱은 임의의 바이트 위치에서 시작하는 작은 데이터 청크를 읽습니다(FileReader API 사용). 해당 데이터 청크에서 JavaScript를 사용하여 기본 문자열 조작을 수행하고 관련 메트릭을 계산합니다. 그러한 지표 중 하나는 DNA 조각을 따라 각 위치에서 일반적으로 보는 A, C, G 및 T의 수를 추적하는 데 도움이 됩니다.
해당 데이터 청크에 대한 메트릭이 계산되면 Plotly.js와 대화식으로 결과를 플롯하고 파일의 다음 청크로 이동합니다. 파일을 작은 청크로 처리하는 이유는 단순히 사용자 경험을 개선하기 위한 것입니다. FASTQ 파일은 일반적으로 수백 기가바이트이기 때문에 전체 파일을 한 번에 처리하는 것은 너무 오래 걸립니다. 0.5MB에서 1MB 사이의 청크 크기는 응용 프로그램을 보다 원활하게 만들고 사용자에게 더 빨리 정보를 반환하지만 이 숫자는 응용 프로그램의 세부 정보와 계산의 양에 따라 달라집니다.
원래 JavaScript 구현의 아키텍처는 상당히 간단했습니다.
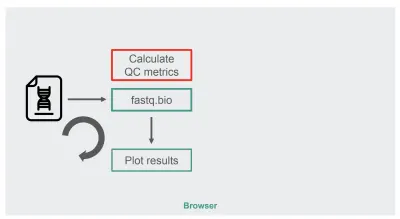
빨간색 상자는 메트릭을 생성하기 위해 문자열 조작을 수행하는 곳입니다. 이 상자는 응용 프로그램에서 더 계산 집약적인 부분이므로 당연히 WebAssembly를 사용한 런타임 최적화의 좋은 후보가 되었습니다.
fastq.bio: WebAssembly 구현
WebAssembly를 활용하여 웹 앱의 속도를 높일 수 있는지 알아보기 위해 FASTQ 파일에 대한 QC 메트릭을 계산하는 기성 도구를 검색했습니다. 특히, 우리는 WebAssembly로 포팅할 수 있도록 C/C++/Rust로 작성된 도구를 찾았고 과학 커뮤니티에서 이미 검증되고 신뢰할 수 있는 도구를 찾았습니다.
약간의 연구 끝에 우리는 시퀀싱 데이터의 품질을 평가하는 데 도움이 될 수 있는 C로 작성된 일반적으로 사용되는 오픈 소스 도구인 seqtk를 사용하기로 결정했습니다(더 일반적으로 해당 데이터 파일을 조작하는 데 사용됨).
WebAssembly로 컴파일하기 전에 먼저 seqtk를 일반적으로 명령줄에서 실행하기 위해 바이너리로 컴파일하는 방법을 고려해 보겠습니다. Makefile에 따르면 다음은 필요한 gcc 주문입니다.
# Compile to binary $ gcc seqtk.c \ -o seqtk \ -O2 \ -lm \ -lz반면에 seqtk를 WebAssembly로 컴파일하기 위해 기존 빌드 도구에 대한 드롭인 교체를 제공하여 WebAssembly에서 더 쉽게 작업할 수 있는 Emscripten 도구 체인을 사용할 수 있습니다. Emscripten이 설치되어 있지 않은 경우 필요한 도구가 포함된 Dockerhub에서 준비한 도커 이미지를 다운로드할 수 있습니다(처음부터 설치할 수도 있지만 일반적으로 시간이 소요됨).

$ docker pull robertaboukhalil/emsdk:1.38.26 $ docker run -dt --name wasm-seqtk robertaboukhalil/emsdk:1.38.26 컨테이너 내부에서 emcc 컴파일러를 gcc 대신 사용할 수 있습니다.
# Compile to WebAssembly $ emcc seqtk.c \ -o seqtk.js \ -O2 \ -lm \ -s USE_ZLIB=1 \ -s FORCE_FILESYSTEM=1보시다시피 바이너리로 컴파일하는 것과 WebAssembly로 컴파일하는 것의 차이점은 최소화됩니다.
- 출력이 바이너리 파일
seqtk가 되는 대신, 우리는 Emscripten에 WebAssembly 모듈의 인스턴스화를 처리하는.wasm및.js를 생성하도록 요청합니다. - zlib 라이브러리를 지원하기 위해
USE_ZLIB플래그를 사용합니다. zlib는 너무 일반적이어서 이미 WebAssembly로 이식되었으며 Emscripten은 이를 우리 프로젝트에 포함할 것입니다. - Emscripten의 가상 파일 시스템은 POSIX와 유사한 파일 시스템(소스 코드는 여기)을 활성화합니다. 단, 브라우저 내의 RAM에서 실행되고 페이지를 새로 고칠 때 사라집니다(IndexedDB를 사용하여 브라우저에 상태를 저장하지 않는 한). 다른 기사).
왜 가상 파일 시스템인가? 이에 대한 답을 얻기 위해 명령줄에서 seqtk를 호출하는 방법과 JavaScript를 사용하여 컴파일된 WebAssembly 모듈을 호출하는 방법을 비교해 보겠습니다.
# On the command line $ ./seqtk fqchk data.fastq # In the browser console > Module.callMain(["fqchk", "data.fastq"]) 가상 파일 시스템에 액세스할 수 있다는 것은 파일 경로 대신 문자열 입력을 처리하기 위해 seqtk를 다시 작성할 필요가 없다는 것을 의미하기 때문에 강력합니다. 가상 파일 시스템에서 데이터 청크를 파일 data.fastq 로 마운트하고 간단히 seqtk의 main() 함수를 호출할 수 있습니다.
WebAssembly로 컴파일된 seqtk를 사용하면 다음과 같은 새로운 fastq.bio 아키텍처가 있습니다.
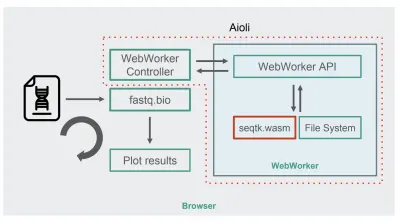
다이어그램에서 볼 수 있듯이 브라우저의 기본 스레드에서 계산을 실행하는 대신 WebWorkers를 사용하여 백그라운드 스레드에서 계산을 실행하고 브라우저의 응답성에 부정적인 영향을 미치지 않도록 합니다. 특히 WebWorker 컨트롤러는 Worker를 시작하고 메인 스레드와의 통신을 관리합니다. 작업자 측에서 API는 수신한 요청을 실행합니다.
그런 다음 작업자에게 방금 마운트한 파일에서 seqtk 명령을 실행하도록 요청할 수 있습니다. seqtk 실행이 완료되면 작업자는 Promise를 통해 결과를 다시 메인 스레드로 보냅니다. 메시지를 받으면 메인 스레드는 결과 출력을 사용하여 차트를 업데이트합니다. JavaScript 버전과 유사하게 파일을 청크로 처리하고 각 반복에서 시각화를 업데이트합니다.
성능 최적화
WebAssembly 사용이 효과가 있었는지 평가하기 위해 초당 처리할 수 있는 읽기 수 측정항목을 사용하여 JavaScript와 WebAssembly 구현을 비교합니다. 두 구현 모두 해당 목적으로 JavaScript를 사용하기 때문에 대화형 그래프를 생성하는 데 걸리는 시간을 무시합니다.
기본적으로 이미 ~9배의 속도 향상을 볼 수 있습니다.
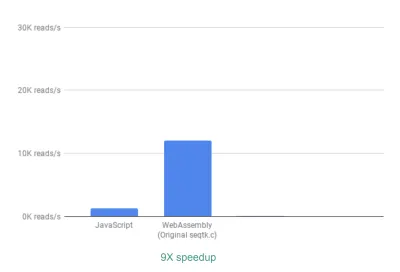
이것은 달성하기가 비교적 쉬웠다는 점을 감안할 때 이미 매우 훌륭합니다(즉, WebAssembly를 이해하면!).
다음으로, seqtk가 일반적으로 유용한 QC 메트릭을 많이 출력하지만 이러한 메트릭 중 많은 부분이 앱에서 실제로 사용되거나 그래프로 표시되지 않는다는 것을 알았습니다. 필요하지 않은 메트릭에 대한 일부 출력을 제거하여 13배의 훨씬 더 빠른 속도 향상을 볼 수 있었습니다.
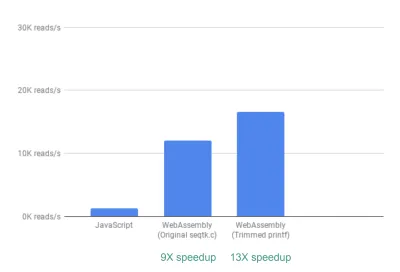
이것은 필요하지 않은 printf 문을 문자 그대로 주석 처리하여 달성하기가 얼마나 쉬운지를 감안할 때 다시 큰 개선입니다.
마지막으로 우리가 조사한 개선 사항이 하나 더 있습니다. 지금까지 fastq.bio가 관심 있는 메트릭을 얻는 방법은 각각 다른 메트릭 세트를 계산하는 두 개의 서로 다른 C 함수를 호출하는 것입니다. 특히, 한 함수는 히스토그램(즉, 범위로 묶은 값 목록)의 형태로 정보를 반환하는 반면, 다른 함수는 DNA 서열 위치의 함수로 정보를 반환합니다. 불행히도 이것은 파일의 동일한 청크를 두 번 읽는다는 것을 의미하므로 불필요합니다.
그래서 우리는 두 함수에 대한 코드를 비록 지저분하긴 하지만 하나의 함수로 병합했습니다(내 C를 다듬을 필요도 없이!). 두 출력의 열 수가 다르기 때문에 JavaScript 측에서 두 가지를 풀기 위해 약간의 랭글링을 수행했습니다. 하지만 그만한 가치가 있었습니다. 그렇게 함으로써 20배 이상의 속도 향상을 달성할 수 있었습니다!
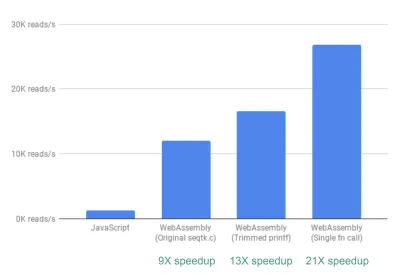
주의 말씀
지금은 경고를 위한 좋은 시간이 될 것입니다. WebAssembly를 사용할 때 항상 20배의 속도 향상을 기대하지 마십시오. 2배 속도 향상 또는 20% 속도 향상만 얻을 수 있습니다. 또는 메모리에 매우 큰 파일을 로드하거나 WebAssembly와 JavaScript 간에 많은 통신이 필요한 경우 속도가 느려질 수 있습니다.
결론
요컨대, 느린 JavaScript 계산을 컴파일된 WebAssembly에 대한 호출로 대체하면 상당한 속도 향상으로 이어질 수 있음을 확인했습니다. 이러한 계산에 필요한 코드는 이미 C에 존재하므로 신뢰할 수 있는 도구를 재사용할 수 있다는 이점이 있습니다. 또한 언급했듯이 WebAssembly가 항상 작업에 적합한 도구는 아니므로( 헐떡임 ! ) 현명하게 사용하십시오.
추가 읽기
- "WebAssembly로 레벨 업", Robert Aboukhalil
WebAssembly 애플리케이션 구축에 대한 실용적인 가이드입니다. - 아이올리(GitHub)
빠른 유전체학 웹 도구를 구축하기 위한 프레임워크입니다. - fastq.bio 소스 코드(GitHub)
DNA 시퀀싱 데이터의 품질 관리를 위한 대화형 웹 도구입니다. - "WebAssembly에 대한 간략한 만화 소개", Lin Clark