
Comment nous avons utilisé WebAssembly pour accélérer notre application Web de 20 fois (étude de cas)
Publié: 2022-03-10Si vous ne l'avez pas entendu, voici le TL; DR : WebAssembly est un nouveau langage qui s'exécute dans le navigateur aux côtés de JavaScript. Oui c'est vrai. JavaScript n'est plus le seul langage qui s'exécute dans le navigateur !
Mais au-delà du simple fait d'être "pas JavaScript", son facteur distinctif est que vous pouvez compiler du code à partir de langages tels que C/C++/Rust ( et plus ! ) vers WebAssembly et les exécuter dans le navigateur. Parce que WebAssembly est typé statiquement, utilise une mémoire linéaire et est stocké dans un format binaire compact, il est également très rapide et pourrait éventuellement nous permettre d'exécuter du code à des vitesses "quasi natives", c'est-à-dire à des vitesses proches de ce que vous d get en exécutant le binaire sur la ligne de commande. La possibilité d'exploiter les outils et les bibliothèques existants pour une utilisation dans le navigateur et le potentiel d'accélération associé sont deux raisons qui rendent WebAssembly si attrayant pour le Web.
Jusqu'à présent, WebAssembly a été utilisé pour toutes sortes d'applications, allant des jeux (par exemple Doom 3) au portage d'applications de bureau sur le Web (par exemple Autocad et Figma). Il est même utilisé en dehors du navigateur, par exemple comme langage efficace et flexible pour l'informatique sans serveur.
Cet article est une étude de cas sur l'utilisation de WebAssembly pour accélérer un outil Web d'analyse de données. À cette fin, nous allons prendre un outil existant écrit en C qui effectue les mêmes calculs, le compiler en WebAssembly et l'utiliser pour remplacer les calculs JavaScript lents.
Remarque : cet article aborde des sujets avancés tels que la compilation de code C, mais ne vous inquiétez pas si vous n'avez pas d'expérience dans ce domaine ; vous pourrez toujours suivre et avoir une idée de ce qui est possible avec WebAssembly.
Contexte
L'application Web avec laquelle nous travaillerons est fastq.bio, un outil Web interactif qui fournit aux scientifiques un aperçu rapide de la qualité de leurs données de séquençage d'ADN ; le séquençage est le processus par lequel nous lisons les « lettres » (c'est-à-dire les nucléotides) dans un échantillon d'ADN.
Voici une capture d'écran de l'application en action :
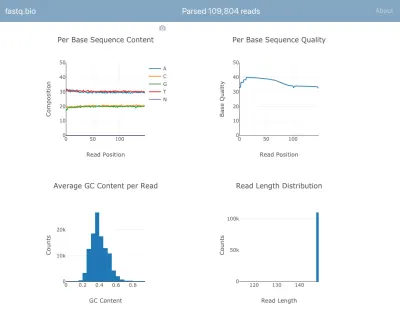
Nous n'entrerons pas dans les détails des calculs, mais en un mot, les graphiques ci-dessus donnent aux scientifiques une idée de la qualité du séquençage et sont utilisés pour identifier en un coup d'œil les problèmes de qualité des données.
Bien qu'il existe des dizaines d'outils de ligne de commande disponibles pour générer de tels rapports de contrôle qualité, l'objectif de fastq.bio est de donner un aperçu interactif de la qualité des données sans quitter le navigateur. Ceci est particulièrement utile pour les scientifiques qui ne sont pas à l'aise avec la ligne de commande.
L'entrée de l'application est un fichier en texte brut qui est produit par l'instrument de séquençage et contient une liste de séquences d'ADN et un score de qualité pour chaque nucléotide dans les séquences d'ADN. Le format de ce fichier est connu sous le nom de "FASTQ", d'où le nom fastq.bio.
Si vous êtes curieux de connaître le format FASTQ (pas nécessaire pour comprendre cet article), consultez la page Wikipedia de FASTQ. (Attention : Le format de fichier FASTQ est connu dans le domaine pour induire des facepalms.)
fastq.bio : l'implémentation de JavaScript
Dans la version originale de fastq.bio, l'utilisateur commence par sélectionner un fichier FASTQ sur son ordinateur. Avec l'objet File , l'application lit un petit bloc de données à partir d'une position d'octet aléatoire (à l'aide de l'API FileReader). Dans ce bloc de données, nous utilisons JavaScript pour effectuer des manipulations de chaînes de base et calculer des métriques pertinentes. Une telle métrique nous aide à suivre le nombre de A, C, G et T que nous voyons généralement à chaque position le long d'un fragment d'ADN.
Une fois les mesures calculées pour ce bloc de données, nous traçons les résultats de manière interactive avec Plotly.js et passons au bloc suivant du fichier. La raison du traitement du fichier en petits morceaux est simplement d'améliorer l'expérience utilisateur : le traitement de l'ensemble du fichier en une seule fois prendrait trop de temps, car les fichiers FASTQ se chiffrent généralement en centaines de gigaoctets. Nous avons constaté qu'une taille de bloc comprise entre 0,5 Mo et 1 Mo rendrait l'application plus transparente et renverrait plus rapidement les informations à l'utilisateur, mais ce nombre variera en fonction des détails de votre application et de la lourdeur des calculs.
L'architecture de notre implémentation JavaScript d'origine était assez simple :
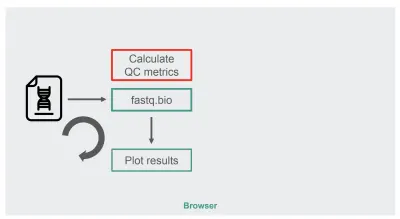
La case en rouge est l'endroit où nous effectuons les manipulations de chaîne pour générer les métriques. Cette boîte est la partie la plus gourmande en calcul de l'application, ce qui en fait naturellement un bon candidat pour l'optimisation de l'exécution avec WebAssembly.
fastq.bio : la mise en œuvre de WebAssembly
Pour déterminer si nous pouvions tirer parti de WebAssembly pour accélérer notre application Web, nous avons recherché un outil prêt à l'emploi qui calcule les métriques QC sur les fichiers FASTQ. Plus précisément, nous avons recherché un outil écrit en C/C++/Rust afin qu'il puisse être porté sur WebAssembly, et qui soit déjà validé et approuvé par la communauté scientifique.
Après quelques recherches, nous avons décidé d'utiliser seqtk, un outil open source couramment utilisé écrit en C qui peut nous aider à évaluer la qualité des données de séquençage (et est plus généralement utilisé pour manipuler ces fichiers de données).
Avant de compiler en WebAssembly, considérons d'abord comment nous compilerions normalement seqtk en binaire pour l'exécuter sur la ligne de commande. Selon le Makefile, voici l'incantation gcc dont vous avez besoin :
# Compile to binary $ gcc seqtk.c \ -o seqtk \ -O2 \ -lm \ -lzD'autre part, pour compiler seqtk en WebAssembly, nous pouvons utiliser la chaîne d'outils Emscripten, qui fournit des remplacements directs pour les outils de construction existants afin de faciliter le travail dans WebAssembly. Si vous n'avez pas installé Emscripten, vous pouvez télécharger une image docker que nous avons préparée sur Dockerhub et qui contient les outils dont vous aurez besoin (vous pouvez également l'installer à partir de zéro, mais cela prend généralement un certain temps) :

$ docker pull robertaboukhalil/emsdk:1.38.26 $ docker run -dt --name wasm-seqtk robertaboukhalil/emsdk:1.38.26 À l'intérieur du conteneur, nous pouvons utiliser le compilateur emcc en remplacement de gcc :
# Compile to WebAssembly $ emcc seqtk.c \ -o seqtk.js \ -O2 \ -lm \ -s USE_ZLIB=1 \ -s FORCE_FILESYSTEM=1Comme vous pouvez le constater, les différences entre la compilation en binaire et WebAssembly sont minimes :
- Au lieu que la sortie soit le fichier binaire
seqtk, nous demandons à Emscripten de générer un.wasmet un.jsqui gère l'instanciation de notre module WebAssembly - Pour prendre en charge la bibliothèque zlib, nous utilisons le drapeau
USE_ZLIB; zlib est si courant qu'il a déjà été porté sur WebAssembly, et Emscripten l'inclura pour nous dans notre projet - Nous activons le système de fichiers virtuel d'Emscripten, qui est un système de fichiers de type POSIX (code source ici), sauf qu'il s'exécute dans la RAM du navigateur et disparaît lorsque vous actualisez la page (sauf si vous enregistrez son état dans le navigateur à l'aide d'IndexedDB, mais c'est pour un autre article).
Pourquoi un système de fichiers virtuel ? Pour répondre à cela, comparons la façon dont nous appellerions seqtk sur la ligne de commande par rapport à l'utilisation de JavaScript pour appeler le module WebAssembly compilé :
# On the command line $ ./seqtk fqchk data.fastq # In the browser console > Module.callMain(["fqchk", "data.fastq"]) Avoir accès à un système de fichiers virtuel est puissant car cela signifie que nous n'avons pas à réécrire seqtk pour gérer les entrées de chaîne au lieu des chemins de fichiers. Nous pouvons monter un bloc de données en tant que fichier data.fastq sur le système de fichiers virtuel et appeler simplement la fonction main() de seqtk dessus.
Avec seqtk compilé sur WebAssembly, voici la nouvelle architecture fastq.bio :
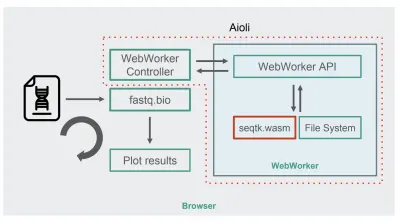
Comme le montre le diagramme, au lieu d'exécuter les calculs dans le thread principal du navigateur, nous utilisons des WebWorkers, qui nous permettent d'exécuter nos calculs dans un thread d'arrière-plan et d'éviter d'affecter négativement la réactivité du navigateur. Concrètement, le contrôleur WebWorker lance le Worker et gère la communication avec le thread principal. Côté Worker, une API exécute les requêtes qu'elle reçoit.
Nous pouvons ensuite demander au Worker d'exécuter une commande seqtk sur le fichier que nous venons de monter. Lorsque seqtk a fini de s'exécuter, le Worker renvoie le résultat au thread principal via une Promise. Une fois qu'il reçoit le message, le thread principal utilise la sortie résultante pour mettre à jour les graphiques. Semblable à la version JavaScript, nous traitons les fichiers en morceaux et mettons à jour les visualisations à chaque itération.
Optimisation des performances
Pour évaluer si l'utilisation de WebAssembly a fait du bien, nous comparons les implémentations JavaScript et WebAssembly en utilisant la métrique du nombre de lectures que nous pouvons traiter par seconde. Nous ignorons le temps nécessaire à la génération de graphiques interactifs, car les deux implémentations utilisent JavaScript à cette fin.
Hors de la boîte, nous voyons déjà une accélération ~9X :
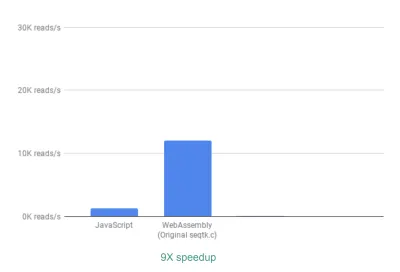
C'est déjà très bien, étant donné que c'était relativement facile à réaliser (c'est-à-dire une fois que vous avez compris WebAssembly !).
Ensuite, nous avons remarqué que bien que seqtk produise de nombreuses métriques QC généralement utiles, bon nombre de ces métriques ne sont pas réellement utilisées ou représentées graphiquement par notre application. En supprimant une partie de la sortie pour les métriques dont nous n'avions pas besoin, nous avons pu voir une accélération encore plus grande de 13X :
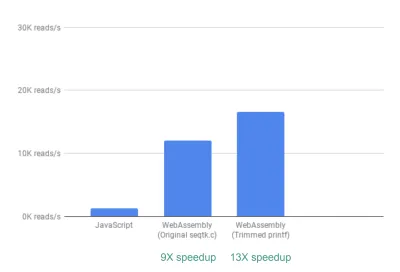
C'est encore une fois une grande amélioration compte tenu de la facilité avec laquelle il a été réalisé, en commentant littéralement les déclarations printf qui n'étaient pas nécessaires.
Enfin, il y a une autre amélioration que nous avons examinée. Jusqu'à présent, fastq.bio obtient les métriques d'intérêt en appelant deux fonctions C différentes, chacune calculant un ensemble différent de métriques. Plus précisément, une fonction renvoie des informations sous la forme d'un histogramme (c'est-à-dire une liste de valeurs que nous regroupons en plages), tandis que l'autre fonction renvoie des informations en fonction de la position de la séquence d'ADN. Malheureusement, cela signifie que le même morceau de fichier est lu deux fois, ce qui est inutile.
Nous avons donc fusionné le code des deux fonctions en une seule fonction, quoique désordonnée (sans même avoir à rafraîchir mon C !). Étant donné que les deux sorties ont des nombres de colonnes différents, nous avons fait quelques querelles du côté JavaScript pour démêler les deux. Mais cela en valait la peine : cela nous a permis d'obtenir une accélération > 20 X !
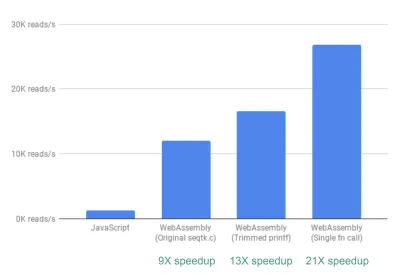
Un mot d'avertissement
Ce serait le bon moment pour une mise en garde. Ne vous attendez pas à toujours obtenir une accélération 20X lorsque vous utilisez WebAssembly. Vous pourriez n'obtenir qu'une accélération 2X ou une accélération de 20%. Ou vous pouvez obtenir un ralentissement si vous chargez des fichiers très volumineux en mémoire ou si vous avez besoin de beaucoup de communication entre le WebAssembly et le JavaScript.
Conclusion
En bref, nous avons vu que le remplacement des calculs JavaScript lents par des appels à WebAssembly compilé peut entraîner des accélérations significatives. Étant donné que le code nécessaire à ces calculs existait déjà en C, nous avons eu l'avantage supplémentaire de réutiliser un outil de confiance. Comme nous l'avons également évoqué, WebAssembly ne sera pas toujours le bon outil pour le travail ( halètement ! ), alors utilisez-le à bon escient.
Lectures complémentaires
- "Passez au niveau supérieur avec WebAssembly", Robert Aboukhalil
Un guide pratique pour créer des applications WebAssembly. - Aïoli (sur GitHub)
Un cadre pour la création d'outils Web de génomique rapide. - code source fastq.bio (sur GitHub)
Un outil Web interactif pour le contrôle de la qualité des données de séquençage de l'ADN. - "Une introduction abrégée de dessin animé à WebAssembly", Lin Clark