
Cómo usamos WebAssembly para acelerar nuestra aplicación web en 20 veces (estudio de caso)
Publicado: 2022-03-10Si no lo ha escuchado, aquí está el TL; DR: WebAssembly es un nuevo lenguaje que se ejecuta en el navegador junto con JavaScript. Sí, eso es correcto. ¡JavaScript ya no es el único lenguaje que se ejecuta en el navegador!
Pero más allá de ser “no JavaScript”, su factor distintivo es que puede compilar código de lenguajes como C/C++/Rust ( ¡y más! ) a WebAssembly y ejecutarlos en el navegador. Debido a que WebAssembly tiene tipado estático, usa una memoria lineal y se almacena en un formato binario compacto, también es muy rápido y eventualmente podría permitirnos ejecutar código a velocidades "casi nativas", es decir, a velocidades cercanas a las que usted necesita. d get ejecutando el binario en la línea de comando. La capacidad de aprovechar las herramientas y bibliotecas existentes para su uso en el navegador y el potencial asociado de aceleración son dos razones que hacen que WebAssembly sea tan atractivo para la web.
Hasta ahora, WebAssembly se ha utilizado para todo tipo de aplicaciones, desde juegos (p. ej., Doom 3) hasta la migración de aplicaciones de escritorio a la web (p. ej., Autocad y Figma). Incluso se usa fuera del navegador, por ejemplo, como un lenguaje eficiente y flexible para la informática sin servidor.
Este artículo es un estudio de caso sobre el uso de WebAssembly para acelerar una herramienta web de análisis de datos. Con ese fin, tomaremos una herramienta existente escrita en C que realiza los mismos cálculos, la compilaremos en WebAssembly y la usaremos para reemplazar los cálculos lentos de JavaScript.
Nota : este artículo profundiza en algunos temas avanzados, como la compilación de código C, pero no se preocupe si no tiene experiencia con eso; aún podrá seguirlo y tener una idea de lo que es posible con WebAssembly.
Antecedentes
La aplicación web con la que trabajaremos es fastq.bio, una herramienta web interactiva que brinda a los científicos una vista previa rápida de la calidad de sus datos de secuenciación de ADN; la secuenciación es el proceso mediante el cual leemos las "letras" (es decir, los nucleótidos) en una muestra de ADN.
Aquí hay una captura de pantalla de la aplicación en acción:
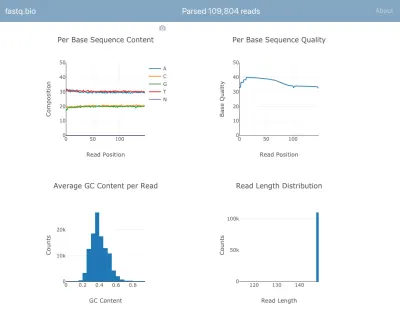
No entraremos en los detalles de los cálculos, pero en pocas palabras, los gráficos anteriores brindan a los científicos una idea de qué tan bien funcionó la secuencia y se utilizan para identificar problemas de calidad de datos de un vistazo.
Aunque hay docenas de herramientas de línea de comandos disponibles para generar dichos informes de control de calidad, el objetivo de fastq.bio es brindar una vista previa interactiva de la calidad de los datos sin salir del navegador. Esto es especialmente útil para los científicos que no se sienten cómodos con la línea de comandos.
La entrada a la aplicación es un archivo de texto sin formato que genera el instrumento de secuenciación y contiene una lista de secuencias de ADN y un puntaje de calidad para cada nucleótido en las secuencias de ADN. El formato de ese archivo se conoce como “FASTQ”, de ahí el nombre fastq.bio.
Si tiene curiosidad sobre el formato FASTQ (no es necesario para comprender este artículo), consulte la página de Wikipedia para FASTQ. (Advertencia: el formato de archivo FASTQ es conocido en el campo para inducir facepalms).
fastq.bio: la implementación de JavaScript
En la versión original de fastq.bio, el usuario comienza seleccionando un archivo FASTQ de su computadora. Con el objeto File , la aplicación lee una pequeña parte de los datos que comienzan en una posición de byte aleatoria (usando la API de FileReader). En esa porción de datos, usamos JavaScript para realizar manipulaciones básicas de cadenas y calcular métricas relevantes. Una de esas métricas nos ayuda a rastrear cuántas A, C, G y T vemos típicamente en cada posición a lo largo de un fragmento de ADN.
Una vez que se calculan las métricas para ese fragmento de datos, trazamos los resultados de forma interactiva con Plotly.js y pasamos al siguiente fragmento del archivo. La razón para procesar el archivo en pequeños fragmentos es simplemente mejorar la experiencia del usuario: procesar todo el archivo a la vez llevaría demasiado tiempo, porque los archivos FASTQ generalmente tienen cientos de gigabytes. Descubrimos que un tamaño de fragmento entre 0,5 MB y 1 MB haría que la aplicación fuera más fluida y devolvería la información al usuario más rápidamente, pero este número variará según los detalles de su aplicación y la cantidad de cálculos.
La arquitectura de nuestra implementación original de JavaScript fue bastante sencilla:
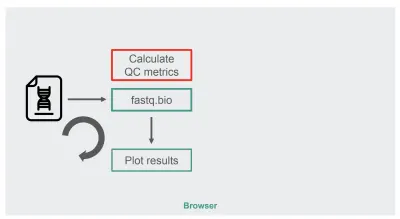
El cuadro en rojo es donde hacemos las manipulaciones de cadenas para generar las métricas. Ese cuadro es la parte de la aplicación que requiere más cómputo, lo que naturalmente lo convirtió en un buen candidato para la optimización del tiempo de ejecución con WebAssembly.
fastq.bio: La implementación de WebAssembly
Para explorar si podíamos aprovechar WebAssembly para acelerar nuestra aplicación web, buscamos una herramienta lista para usar que calcula las métricas de control de calidad en archivos FASTQ. Específicamente, buscamos una herramienta escrita en C/C++/Rust para que pudiera migrarse a WebAssembly, y una que ya estuviera validada y en la que confiara la comunidad científica.
Después de algunas investigaciones, decidimos usar seqtk, una herramienta de código abierto de uso común escrita en C que puede ayudarnos a evaluar la calidad de los datos de secuenciación (y se usa más generalmente para manipular esos archivos de datos).
Antes de compilar en WebAssembly, primero consideremos cómo compilaríamos normalmente seqtk en binario para ejecutarlo en la línea de comandos. Según el Makefile, este es el conjuro gcc que necesitas:
# Compile to binary $ gcc seqtk.c \ -o seqtk \ -O2 \ -lm \ -lzPor otro lado, para compilar seqtk en WebAssembly, podemos usar la cadena de herramientas Emscripten, que proporciona reemplazos directos para las herramientas de compilación existentes para facilitar el trabajo en WebAssembly. Si no tiene Emscripten instalado, puede descargar una imagen acoplable que preparamos en Dockerhub que tiene las herramientas que necesitará (también puede instalarla desde cero, pero eso generalmente toma un tiempo):

$ docker pull robertaboukhalil/emsdk:1.38.26 $ docker run -dt --name wasm-seqtk robertaboukhalil/emsdk:1.38.26 Dentro del contenedor, podemos usar el compilador emcc como reemplazo de gcc :
# Compile to WebAssembly $ emcc seqtk.c \ -o seqtk.js \ -O2 \ -lm \ -s USE_ZLIB=1 \ -s FORCE_FILESYSTEM=1Como puede ver, las diferencias entre compilar binario y WebAssembly son mínimas:
- En lugar de que la salida sea el archivo binario
seqtk, le pedimos a Emscripten que genere un.wasmy un.jsque maneje la creación de instancias de nuestro módulo WebAssembly. - Para admitir la biblioteca zlib, usamos la bandera
USE_ZLIB; zlib es tan común que ya ha sido portado a WebAssembly, y Emscripten lo incluirá en nuestro proyecto - Habilitamos el sistema de archivos virtual de Emscripten, que es un sistema de archivos similar a POSIX (código fuente aquí), excepto que se ejecuta en la RAM dentro del navegador y desaparece cuando actualiza la página (a menos que guarde su estado en el navegador usando IndexedDB, pero eso es para otro artículo).
¿Por qué un sistema de archivos virtual? Para responder a eso, comparemos cómo llamaríamos a seqtk en la línea de comando versus usar JavaScript para llamar al módulo WebAssembly compilado:
# On the command line $ ./seqtk fqchk data.fastq # In the browser console > Module.callMain(["fqchk", "data.fastq"]) Tener acceso a un sistema de archivos virtual es poderoso porque significa que no tenemos que volver a escribir seqtk para manejar entradas de cadenas en lugar de rutas de archivos. Podemos montar una porción de datos como el archivo data.fastq en el sistema de archivos virtual y simplemente llamar a la función main() de seqtk.
Con seqtk compilado en WebAssembly, aquí está la nueva arquitectura fastq.bio:
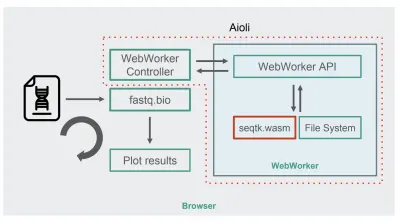
Como se muestra en el diagrama, en lugar de ejecutar los cálculos en el subproceso principal del navegador, usamos WebWorkers, que nos permite ejecutar nuestros cálculos en un subproceso en segundo plano y evita afectar negativamente la capacidad de respuesta del navegador. Específicamente, el controlador de WebWorker inicia Worker y administra la comunicación con el subproceso principal. Del lado del trabajador, una API ejecuta las solicitudes que recibe.
Luego podemos pedirle al Worker que ejecute un comando seqtk en el archivo que acabamos de montar. Cuando seqtk termina de ejecutarse, Worker envía el resultado al subproceso principal a través de una Promesa. Una vez que recibe el mensaje, el hilo principal usa la salida resultante para actualizar los gráficos. Similar a la versión de JavaScript, procesamos los archivos en fragmentos y actualizamos las visualizaciones en cada iteración.
Optimización del rendimiento
Para evaluar si el uso de WebAssembly funcionó, comparamos las implementaciones de JavaScript y WebAssembly utilizando la métrica de cuántas lecturas podemos procesar por segundo. Ignoramos el tiempo que toma generar gráficos interactivos, ya que ambas implementaciones usan JavaScript para ese propósito.
Fuera de la caja, ya vemos una aceleración de ~9X:
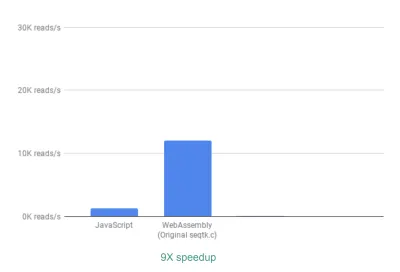
Esto ya es muy bueno, dado que fue relativamente fácil de lograr (¡eso es una vez que entiendes WebAssembly!).
A continuación, notamos que aunque seqtk genera una gran cantidad de métricas de control de calidad generalmente útiles, muchas de estas métricas en realidad no son utilizadas ni graficadas por nuestra aplicación. Al eliminar algunos de los resultados de las métricas que no necesitábamos, pudimos ver una aceleración aún mayor de 13X:
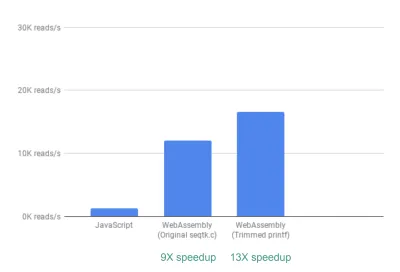
Nuevamente, esto es una gran mejora dado lo fácil que fue lograrlo, al comentar literalmente las declaraciones de printf que no eran necesarias.
Finalmente, hay una mejora más que investigamos. Hasta ahora, la forma en que fastq.bio obtiene las métricas de interés es llamando a dos funciones C diferentes, cada una de las cuales calcula un conjunto diferente de métricas. Específicamente, una función devuelve información en forma de histograma (es decir, una lista de valores que agrupamos en rangos), mientras que la otra función devuelve información en función de la posición de la secuencia de ADN. Desafortunadamente, esto significa que el mismo fragmento de archivo se lee dos veces, lo cual es innecesario.
Así que fusionamos el código de las dos funciones en una función, aunque desordenada (¡sin siquiera tener que repasar mi C!). Dado que las dos salidas tienen diferentes números de columnas, hicimos algunas disputas en el lado de JavaScript para desenredar las dos. Pero valió la pena: hacerlo nos permitió lograr una aceleración de >20X.
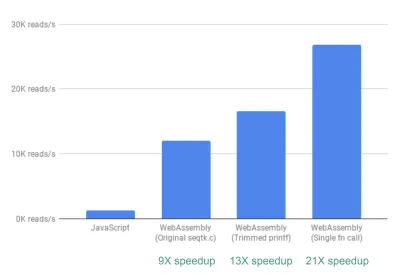
Una palabra de precaución
Ahora sería un buen momento para una advertencia. No espere obtener siempre una aceleración de 20X cuando use WebAssembly. Es posible que solo obtenga una aceleración de 2X o una aceleración del 20%. O puede ralentizarse si carga archivos muy grandes en la memoria o requiere mucha comunicación entre WebAssembly y JavaScript.
Conclusión
En resumen, hemos visto que reemplazar los cálculos lentos de JavaScript con llamadas a WebAssembly compilado puede conducir a aceleraciones significativas. Dado que el código necesario para esos cálculos ya existía en C, obtuvimos el beneficio adicional de reutilizar una herramienta confiable. Como también mencionamos, WebAssembly no siempre será la herramienta adecuada para el trabajo ( ¡jadeo! ), así que utilícela sabiamente.
Otras lecturas
- “Suba de nivel con WebAssembly”, Robert Aboukhalil
Una guía práctica para crear aplicaciones WebAssembly. - Alioli (en GitHub)
Un marco para construir herramientas web de genómica rápida. - código fuente fastq.bio (en GitHub)
Una herramienta web interactiva para el control de calidad de los datos de secuenciación de ADN. - "Una introducción abreviada de dibujos animados a WebAssembly", Lin Clark