
WebAssemblyを使用してWebアプリを20倍高速化した方法(ケーススタディ)
公開: 2022-03-10聞いたことがない方のために、TL; DRをご紹介します。WebAssemblyは、JavaScriptと一緒にブラウザで実行される新しい言語です。 はい、そうです。 ブラウザで実行される言語はJavaScriptだけではなくなりました。
ただし、「JavaScriptではない」というだけでなく、C / C ++ / Rust(およびその他! )などの言語からWebAssemblyにコードをコンパイルして、ブラウザーで実行できるという特徴があります。 WebAssemblyは静的に型指定され、線形メモリを使用し、コンパクトなバイナリ形式で保存されるため、非常に高速であり、最終的には「ネイティブに近い」速度、つまりユーザーに近い速度でコードを実行できるようになる可能性があります。 dコマンドラインでバイナリを実行して取得します。 ブラウザで使用するために既存のツールとライブラリを活用する機能と、それに関連する高速化の可能性が、WebAssemblyをWebにとって非常に魅力的なものにしている2つの理由です。
これまでのところ、WebAssemblyは、ゲーム(Doom 3など)からデスクトップアプリケーションのWebへの移植(AutocadやFigmaなど)まで、あらゆる種類のアプリケーションに使用されてきました。 たとえば、サーバーレスコンピューティング用の効率的で柔軟な言語として、ブラウザの外部でも使用されます。
この記事は、WebAssemblyを使用してデータ分析Webツールを高速化するケーススタディです。 そのために、同じ計算を実行するCで記述された既存のツールを使用して、それをWebAssemblyにコンパイルし、それを使用して低速のJavaScript計算を置き換えます。
注:この記事では、Cコードのコンパイルなどの高度なトピックについて詳しく説明しますが、経験がなくても心配する必要はありません。 それでも、WebAssemblyで何が可能かを理解し、理解することができます。
バックグラウンド
私たちが使用するWebアプリはfastq.bioです。これは、科学者にDNAシーケンシングデータの品質のクイックプレビューを提供するインタラクティブなWebツールです。 シーケンシングは、DNAサンプルの「文字」(つまりヌクレオチド)を読み取るプロセスです。
動作中のアプリケーションのスクリーンショットは次のとおりです。
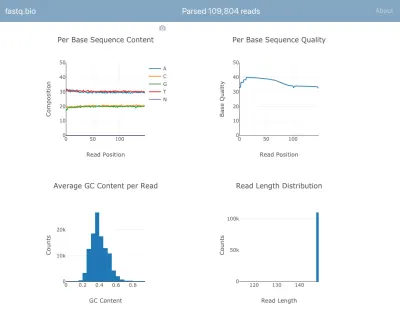
計算の詳細については説明しませんが、簡単に言うと、上記のプロットは、シーケンスがどの程度うまくいったかを科学者に示し、データ品質の問題を一目で特定するために使用されます。
このような品質管理レポートを生成するために利用できるコマンドラインツールは数十ありますが、fastq.bioの目標は、ブラウザーを離れることなくデータ品質のインタラクティブなプレビューを提供することです。 これは、コマンドラインに慣れていない科学者にとって特に便利です。
アプリへの入力は、シーケンス機器によって出力されるプレーンテキストファイルであり、DNAシーケンスのリストとDNAシーケンス内の各ヌクレオチドの品質スコアが含まれています。 そのファイルの形式は「FASTQ」と呼ばれるため、fastq.bioという名前になります。
FASTQ形式(この記事を理解する必要はありません)に興味がある場合は、ウィキペディアのページでFASTQを確認してください。 (警告:FASTQファイル形式は、facepalmsを誘発することが現場で知られています。)
fastq.bio:JavaScriptの実装
fastq.bioの元のバージョンでは、ユーザーは自分のコンピューターからFASTQファイルを選択することから始めます。 Fileオブジェクトを使用すると、アプリはランダムなバイト位置から始まるデータの小さなチャンクを読み取ります(FileReader APIを使用)。 そのデータのチャンクでは、JavaScriptを使用して基本的な文字列操作を実行し、関連するメトリックを計算します。 そのようなメトリックの1つは、DNAフラグメントに沿った各位置で通常見られるA、C、G、およびTの数を追跡するのに役立ちます。
そのデータチャンクのメトリックが計算されたら、Plotly.jsを使用してインタラクティブに結果をプロットし、ファイル内の次のチャンクに移動します。 ファイルを小さなチャンクで処理する理由は、単にユーザーエクスペリエンスを向上させるためです。FASTQファイルは一般に数百ギガバイトであるため、ファイル全体を一度に処理するには時間がかかりすぎます。 0.5MBから1MBのチャンクサイズを使用すると、アプリケーションがよりシームレスになり、ユーザーに情報がより迅速に返されることがわかりましたが、この数値は、アプリケーションの詳細と計算の重さによって異なります。
元のJavaScript実装のアーキテクチャは、かなり単純でした。
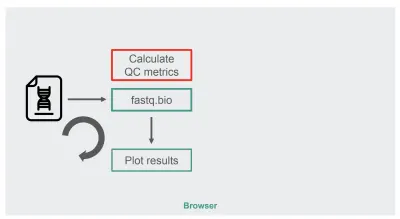
赤いボックスは、文字列操作を行ってメトリックを生成する場所です。 このボックスは、アプリケーションのより計算集約的な部分であり、当然、WebAssemblyを使用したランタイム最適化の候補として適しています。
fastq.bio:WebAssemblyの実装
WebAssemblyを活用してWebアプリを高速化できるかどうかを調べるために、FASTQファイルのQCメトリックを計算する既成のツールを検索しました。 具体的には、C / C ++ / Rustで記述されたツールを探して、WebAssemblyへの移植に対応できるようにしました。また、科学界ですでに検証され、信頼されているツールを探しました。
いくつかの調査の結果、シーケンスデータの品質を評価するのに役立つ(より一般的にはこれらのデータファイルの操作に使用される)Cで記述された一般的に使用されるオープンソースツールであるseqtkを使用することにしました。
WebAssemblyにコンパイルする前に、まず、seqtkをバイナリにコンパイルしてコマンドラインで実行する方法を考えてみましょう。 Makefileによると、これは必要なgccの呪文です。
# Compile to binary $ gcc seqtk.c \ -o seqtk \ -O2 \ -lm \ -lz一方、seqtkをWebAssemblyにコンパイルするには、Emscriptenツールチェーンを使用できます。これは、既存のビルドツールのドロップイン置換を提供して、WebAssemblyでの作業を容易にします。 Emscriptenがインストールされていない場合は、必要なツールを備えたDockerhubで準備したDockerイメージをダウンロードできます(最初からインストールすることもできますが、通常は時間がかかります)。

$ docker pull robertaboukhalil/emsdk:1.38.26 $ docker run -dt --name wasm-seqtk robertaboukhalil/emsdk:1.38.26 コンテナ内では、 gccの代わりにemccコンパイラを使用できます。
# Compile to WebAssembly $ emcc seqtk.c \ -o seqtk.js \ -O2 \ -lm \ -s USE_ZLIB=1 \ -s FORCE_FILESYSTEM=1ご覧のとおり、バイナリへのコンパイルとWebAssemblyへのコンパイルの違いはごくわずかです。
- 出力がバイナリファイル
seqtkである代わりに、EmscriptenにWebAssemblyモジュールのインスタンス化を処理する.wasmと.jsを生成するように依頼します - zlibライブラリをサポートするために、フラグ
USE_ZLIBを使用します。 zlibは非常に一般的であるため、すでにWebAssemblyに移植されており、Emscriptenはそれをプロジェクトに含めます。 - POSIXのようなファイルシステムであるEmscriptenの仮想ファイルシステム(ここではソースコード)を有効にしますが、ブラウザー内のRAMで実行され、ページを更新すると消えます(IndexedDBを使用してブラウザーに状態を保存しない限り)。別の記事のために)。
なぜ仮想ファイルシステムなのか? これに答えるために、コマンドラインでseqtkを呼び出す方法と、JavaScriptを使用してコンパイルされたWebAssemblyモジュールを呼び出す方法を比較してみましょう。
# On the command line $ ./seqtk fqchk data.fastq # In the browser console > Module.callMain(["fqchk", "data.fastq"]) 仮想ファイルシステムにアクセスできることは、ファイルパスの代わりに文字列入力を処理するためにseqtkを書き直す必要がないことを意味するため、強力です。 データのチャンクをファイルdata.fastqとして仮想ファイルシステムにマウントし、その上でseqtkのmain()関数を呼び出すだけです。
seqtkをWebAssemblyにコンパイルすると、新しいfastq.bioアーキテクチャが次のようになります。
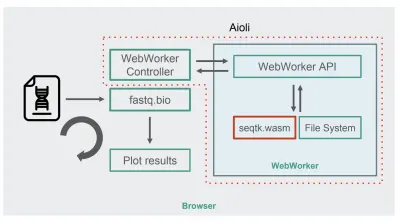
図に示すように、ブラウザーのメインスレッドで計算を実行する代わりに、WebWorkersを使用します。これにより、バックグラウンドスレッドで計算を実行し、ブラウザーの応答性に悪影響を与えないようにすることができます。 具体的には、WebWorkerコントローラーがワーカーを起動し、メインスレッドとの通信を管理します。 ワーカー側では、APIは受信したリクエストを実行します。
次に、マウントしたファイルに対してseqtkコマンドを実行するようにワーカーに要求できます。 seqtkの実行が終了すると、ワーカーは結果をPromise経由でメインスレッドに送り返します。 メッセージを受信すると、メインスレッドは結果の出力を使用してチャートを更新します。 JavaScriptバージョンと同様に、ファイルをチャンクで処理し、反復ごとに視覚化を更新します。
パフォーマンスの最適化
WebAssemblyの使用が効果的かどうかを評価するために、1秒あたりに処理できる読み取り数のメトリックを使用してJavaScriptとWebAssemblyの実装を比較します。 どちらの実装もその目的でJavaScriptを使用しているため、インタラクティブグラフの生成にかかる時間は無視します。
箱から出してすぐに、すでに最大9倍のスピードアップが見られます。
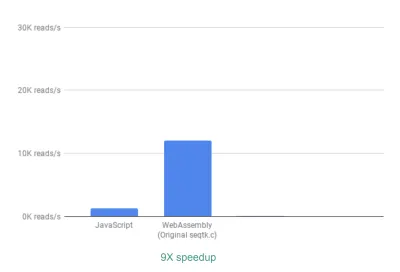
達成するのが比較的簡単だったことを考えると、これはすでに非常に優れています(つまり、WebAssemblyを理解したら!)。
次に、seqtkは一般的に有用なQCメトリックを多数出力しますが、これらのメトリックの多くは実際にはアプリで使用またはグラフ化されていないことに気付きました。 不要なメトリックの出力の一部を削除することで、13倍のさらに高速化を確認できました。
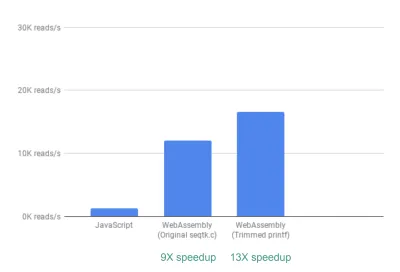
これも、不要なprintfステートメントを文字通りコメントアウトすることで、簡単に達成できることを考えると、大きな改善です。
最後に、調査したもう1つの改善点があります。 これまでのところ、fastq.bioが対象のメトリックを取得する方法は、それぞれが異なるメトリックのセットを計算する2つの異なるC関数を呼び出すことです。 具体的には、一方の関数はヒストグラムの形式で情報(つまり、範囲にビン化する値のリスト)を返しますが、もう一方の関数はDNA配列の位置の関数として情報を返します。 残念ながら、これはファイルの同じチャンクが2回読み取られることを意味し、これは不要です。
そこで、2つの関数のコードを1つの関数にマージしました(厄介ではありますが)(Cをブラッシュアップする必要さえありません!)。 2つの出力の列数が異なるため、JavaScript側でラングリングを行って2つを解きほぐしました。 しかし、それだけの価値はありました。そうすることで、20倍以上の高速化を実現できました。
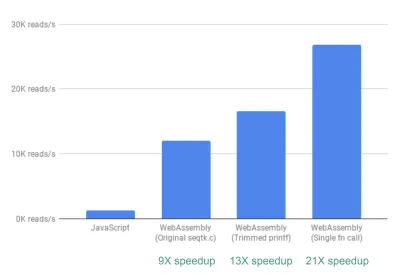
注意の言葉
今は警告のための良い時期でしょう。 WebAssemblyを使用するときに、常に20倍のスピードアップを期待しないでください。 2倍のスピードアップまたは20%のスピードアップしか得られない可能性があります。 または、メモリに非常に大きなファイルをロードしたり、WebAssemblyとJavaScriptの間で多くの通信が必要になったりすると、速度が低下する可能性があります。
結論
つまり、低速のJavaScript計算をコンパイル済みWebAssemblyの呼び出しに置き換えると、大幅な高速化につながる可能性があることがわかりました。 これらの計算に必要なコードはすでにCに存在していたため、信頼できるツールを再利用できるという追加のメリットがありました。 また触れたように、WebAssemblyは必ずしもその仕事に適したツールではないので(あえぎ! )、賢明に使用してください。
参考文献
- 「WebAssemblyでレベルアップ」、Robert Aboukhalil
WebAssemblyアプリケーションを構築するための実用的なガイド。 - Aioli(GitHub上)
高速ゲノミクスWebツールを構築するためのフレームワーク。 - fastq.bioソースコード(GitHub上)
DNAシーケンシングデータの品質管理のためのインタラクティブなウェブツール。 - 「WebAssemblyの要約漫画紹介」LinClark